
Latest Issue
Just Accepted Online First List of Issues Document Types
Issue 12, 2024
REVIEW
2024, 42(12): 1855-1880. DOI: 10.1007/s10118-024-3203-8Published(online): 2024-12-03Abstract Full text L-PDFAbstract

RESEARCH ARTICLE
2024, 42(12): 1881-1887. DOI: 10.1007/s10118-024-3181-xPublished(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 1888-1896. DOI: 10.1007/s10118-024-3225-2Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 1897-1904. DOI: 10.1007/s10118-024-3217-2Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 1905-1914. DOI: 10.1007/s10118-024-3235-0Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 1915-1924. DOI: 10.1007/s10118-024-3223-4Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 1925-1932. DOI: 10.1007/s10118-024-3193-6Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 1933-1940. DOI: 10.1007/s10118-024-3188-3Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 1941-1947. DOI: 10.1007/s10118-024-3231-4Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 1948-1956. DOI: 10.1007/s10118-024-3229-yPublished(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 1957-1965. DOI: 10.1007/s10118-024-3218-1Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 1966-1975. DOI: 10.1007/s10118-024-3212-7Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 1976-1985. DOI: 10.1007/s10118-024-3219-0Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
Scalable and High-Quality Monolayer Graphene Transfer onto Polymer Membranes Assisted by Camphor Enhanced Publication
2024, 42(12): 1986-2001. DOI: 10.1007/s10118-024-3207-4Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 2002-2010. DOI: 10.1007/s10118-024-3201-xPublished(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 2011-2020. DOI: 10.1007/s10118-024-3240-3Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 2021-2029. DOI: 10.1007/s10118-024-3170-0Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 2030-2037. DOI: 10.1007/s10118-024-3194-5Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 2038-2047. DOI: 10.1007/s10118-024-3216-3Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 2048-2058. DOI: 10.1007/s10118-024-3215-4Published(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2024, 42(12): 2059-2068. DOI: 10.1007/s10118-024-3237-yPublished(online): 2024-12-03Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
Interplay between Hydrogen Bond Network and Entangled Network in Polymers During Stretching Based on Molecular Simulations Enhanced Publication
2024, 42(12): 2069-2080. DOI: 10.1007/s10118-024-3227-0Published(online): 2024-12-03Abstract Full text L-PDFAbstract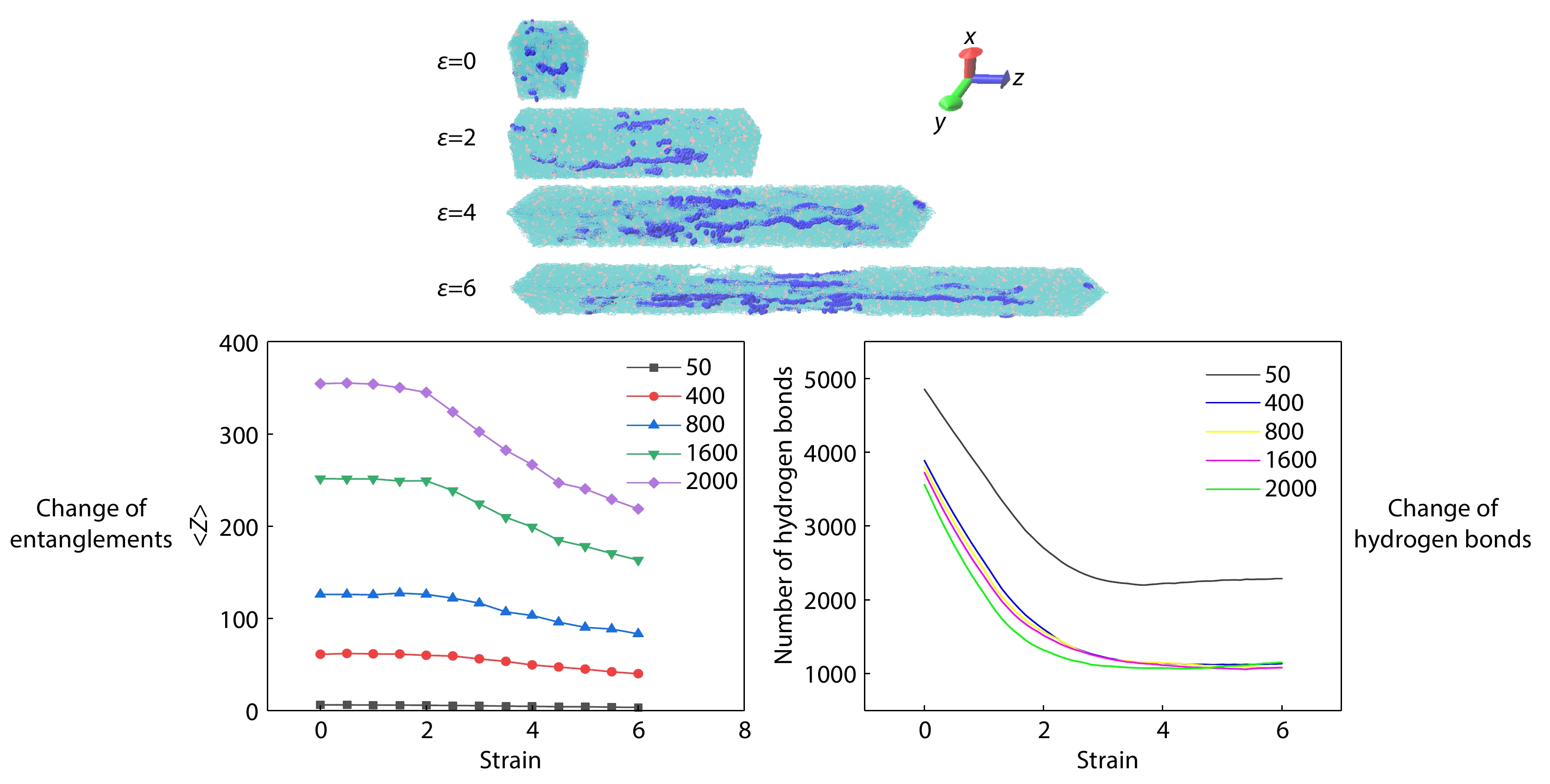
- Technical support is provided by Beijing Founder electronics co., LTD 京ICP备09064830号-19
 京公网安备11010802024621
京公网安备11010802024621 - It is recommended to read the content of this site in Chrome&IE9+. Please switch to extreme mode in browser 360.
- Cookies We use cookies to help provide and enhance our service and tailor content. By continuing, you agree to the use of cookies.
0